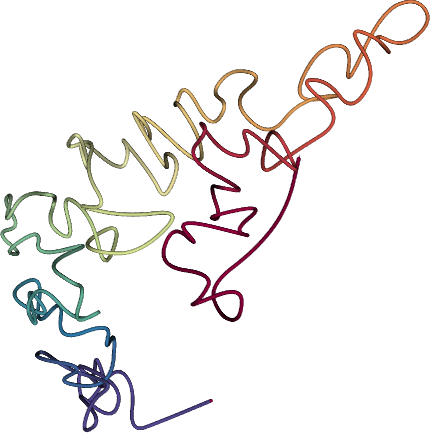
Recent technological advances allow the measurement, in a single Hi-C experiment, of the frequencies of physical contacts among pairs of genomic loci at a genome-wide scale. The next challenge is to infer, from the resulting DNA-DNA contact maps, accurate three dimensional models of how chromosomes fold and fit into the nucleus. Many existing inference methods rely upon multidimensional scaling (MDS), in which the pairwise distances of the inferred model are optimized to resemble pairwise distances derived directly from the contact counts. These approaches, however, often optimize a heuristic objective function and require strong assumptions about the biophysics of DNA to transform interaction frequencies to spatial distance, and thereby may lead to incorrect structure reconstruction.
In pastis, we propose a novel approach to infer a consensus three- dimensional structure of a genome from Hi-C data. The method incorporates a statistical model of the contact counts, assuming that the counts between two loci follow a Poisson distribution whose intensity decreases with the physical distances between the loci. The method can automatically adjust the transfer function relating the spatial distance to the Poisson intensity and infer a genome structure that best explains the observed data.
The package pastis contains four methods to infer the three dimensional methods of a genome from Hi-C data: MDS, NMDS, PM1, PM2. MDS and NMDS are algorithms from the multidimensional scaling family, while PM1 and PM2 are novel approaches, derived from a statistical modeling of the interaction counts and the physical distances.
Download the latest version of pastis here or fork the code on github.
N. Varoquaux, F. Ay, W. S. Noble, and J.-P. Vert. A statistical approach for inferring the 3D structure of the genome. <http://bioinformatics.oxfordjournals.org/content/30/12/i26.short> Bioinformatics, 30(12):i26–i33, 2014.
A. G. Cauer, G. Yardimci, J.-P. Vert, N. Varoquaux, and W. S. Noble. Inferring diploid 3D chromatin structures from Hi-C data. <http://drops.dagstuhl.de/opus/volltexte/2019/11041/> In 19th International Workshop on Algorithms in Bioinformatics (WABI 2019), volume 143 of Leibniz International Proceedings in Informatics (LIPIcs), pages 11:1–11:13, Dagstuhl, Germany, 2019. Schloss Dagstuhl–Leibniz-Zentrum fuer Informatik.


