Modern technologies like DNA microarrays or high-throughput sequencing are revolutionising biology and medical research. By allowing the collection of large amounts of measures at the molecular level on living organisms, they pave the way to a quantitative and rationale analysis of biological systems. Unsurprisingly, statistics and machine learning play an important role in this revolution. By processing large collections of datasets, they allow to extract new biological knowledge and infer predictive models.
The goal of this course is to present a few modern statistical learning techniques, and to touch upon a selected panel of applications in computational and systems biology. We will study in particular support vector machines (SVM) and kernels, as well as feature selection techniques including lasso regression. Applications include protein annotation, virtual screening in drug design, prognostic and predictive models for personalised medicine in oncology, and gene network inference in systems biology.
| Date | Topic |
| Tuesday, Feb 11 | Introduction, learning in high dimension |
| Tuesday, Feb 25 | SVM |
| Tuesday, Mar 4 | Kernels for strings and graphs; lasso |
| Tuesday, Mar 11 | Group lasso, fused lasso, structured sparsity |
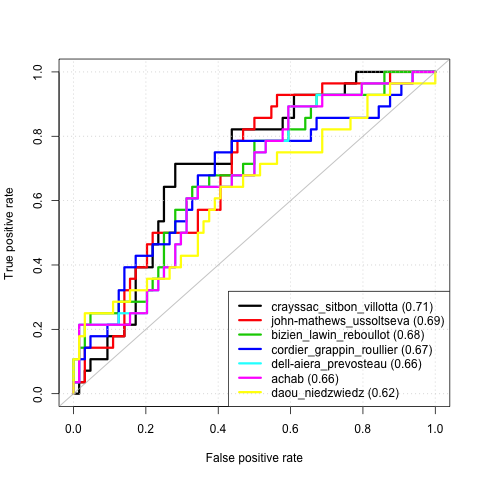
We have collected gene expression levels for 12065 genes on 184 early-stage breast cancer samples: xtrain.txt (each row is a gene, each column a sample; the first element of each row is the gene ID). After surgical removal of the tumour, some unfortunately relapsed within 5 years (label=+1), while other did not (label=-1). The labels of the the 184 samples are available in the file ytrain.txt. Our goal is to design a predictive model in order to predict whether a tumour is likely to relapse or not, from its gene expression values. If we can predict the risk of relapse accurately, then we may be able to adapt the treatment given to the patient, e.g., limiting chemotherapy if the risk is low, or increasing it if it is high.
In addition, we know that genes interact with each other in the cell. We have collected in the file ppi.txt a set of 430815 known interactions between genes. Each row gives the IDs of two genes know to interact.
Please send a short report and your predictions to jean-philippe.vert@mines.org before April 15, 2014. The predictions will be scored in terms of area under the ROC curve (AUC).
You may do this project alone or in groups of 2 or 3 students (encouraged).